Research.
Research Overview:
Cancer Drivers:
Current cancer genomics databases have accumulated millions of somatic mutations that remain to be further explored. Due to the over-excess mutations unrelated to cancer, the great challenge is to identify somatic mutations that are cancer-driven. Under the notion that carcinogenesis is a form of somatic-cell evolution, we developed a software CanDriS (Cancer Driver Sites) based on two-component mixture model: while the ground component corresponds to passenger mutations, the rapidly-evolving component corresponds to driver mutations.
Neoantigen:
Neoantigens have been acknowledged as ideal targets for cancer immunotherapies, such as cancer vaccines and T-cell immunotherapies. Recent studies also indicated that neoantigens are closely related to the therapeutic effect of immune checkpoint blockade therapies. Because of the large workload of experimental verification, the practical solution is to take advantage of bioinformatics. We developed the neoantigen prediction method DeepHLApan, one-stop neoantigen predition tool TSNAD, and neoantigen database TSNAdb for better clinical usage of neoantigen.
COVID19:
Since the outbreak of Coronavirus Disease 2019 (COVID-19) in late December 2019, it has brought significant harm and challenges to over 200 countries and regions around the world. Considering the seriousness of the recent outbreaks of zoonotic coronaviruses, therapeutic agents and vaccines for pan-coronaviruses should be developed to cope with the hCoV outbreaks in the present and in the future. Here, we predict all the potential B/T cell epitopes for SARS-CoV, 2019-nCoV, and MERS-CoV, and develop the Coronavirus Immuno-Epitope databse COVIEdb, to provide potential targets for pan-coronaviruses vaccine development. RaTG13-CoV is included because of its high homology with 2019-nCoV (96% whole genome identity).
Software
TSNAD
v1.0
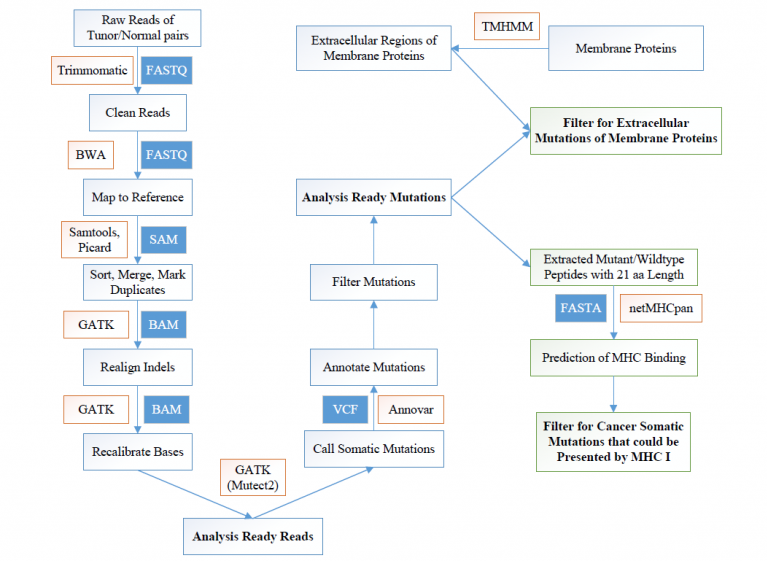
Description:
We develop an integrated software under the Linux operation system called TSNAD through calling a series of the-state-of-art software or tools. It is completely automated and user-friendly framework, which is mainly designed for users who have little programming experience. It consists of two toolkits, mutation detection and antigen predictiion. It aims to detect somatic mutation and predict potential tumor-specific mutated antigens. Each toolkit is a two-step process: (1) configure parameters, (2) run corresponding toolkit.
v2.0
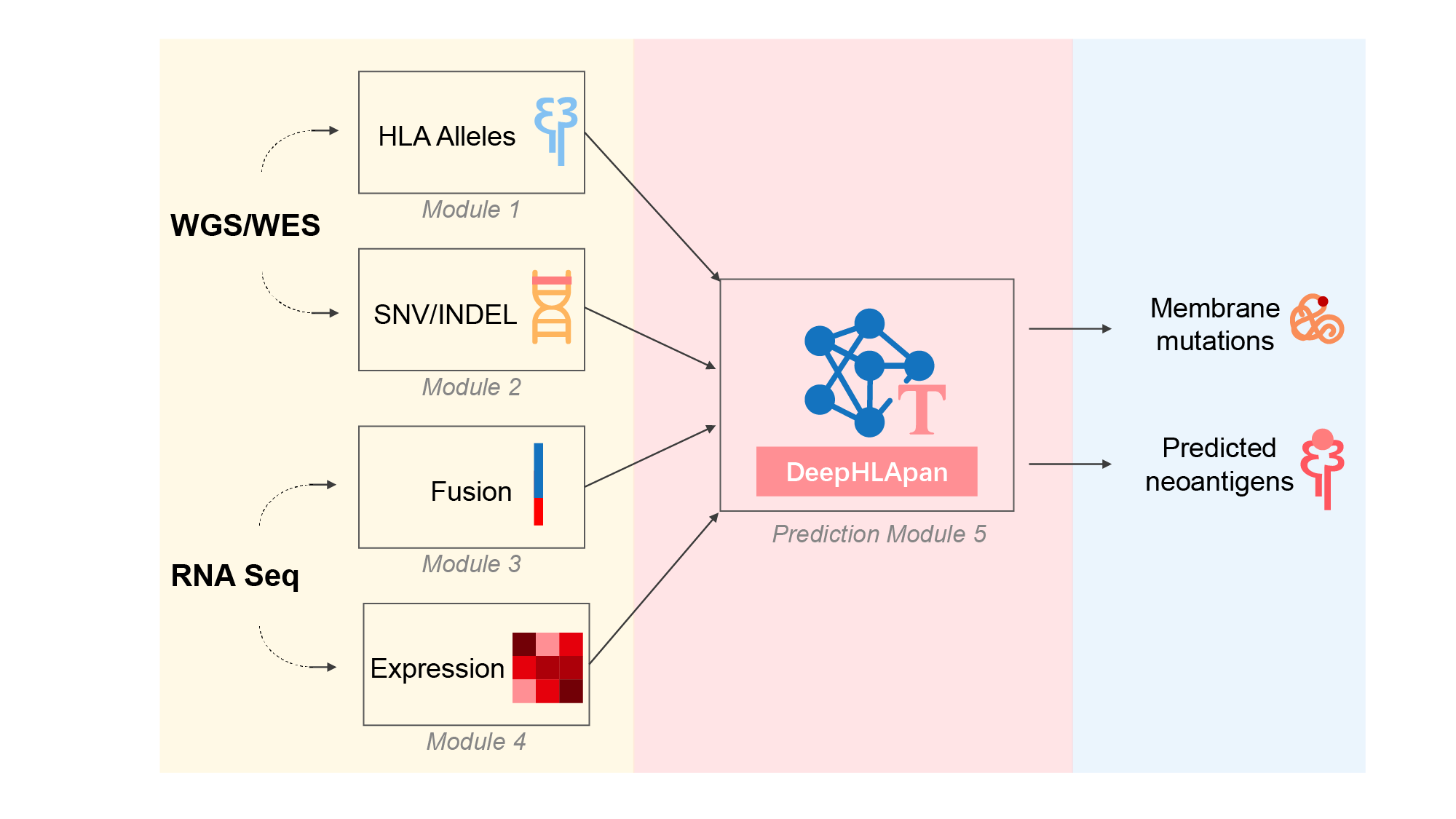
Description:
we present an updated version of TSNAD that implements new features and improvements including: (i) update all the embedded tools into the latest version, (ii) add the function of RNA-Seq data analysis including gene expression and gene fusion analyses, (iii) support both GRCh38 and GRCh37 version of reference genome when calling mutations, (iv) add the neoantigen prediction derived from INDELs and gene fusions, besides SNVs, (v) replace NetMHCpan with our developed tool DeepHLApan and provide a web service of TSNAD, (vi) provide the installation method of Docker which comprises all the needed tools and reference files. TSNAD v2.0 achieves high performance on the standard dataset that the Tumor Neoantigen Selection Alliance (TESLA) provided. TSNAD v2.0 is implemented in Perl and Python. With an installed Docker, TSNAD v2.0 could be used on any operation systems including Linux and Windows.
Publication(s):
1. Zhou Z#,*, Wu J#, Ren J, Chen W, Zhao W, Gu X, Chi Y, He Q, Yang B, Wu J*, Chen S*. TSNAD v2.0: A one-stop software solution for tumor-specific neoantigen detection. Comput Struct Biotechnol J, 2021, 19:4510-4516.2. Zhou Z#, Lyu X#, Wu J, Yang X, Wu S, Zhou J, Gu X, Su Z*, Chen S*. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R Soc Open Sci. 2017, 4: 170050.
DeepHLApan
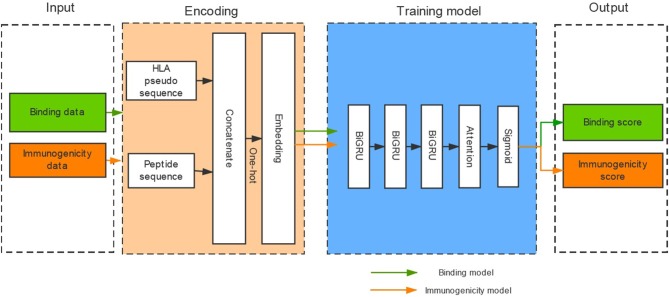
Description:
We developed a novel software called DeepHLApan that can predict peptide binding affinity accurately. We designed this software based on the recurrent neural networks (RNNs) and trained it with a large dataset containing 335,102 peptide-HLA pairs which allow us to predict peptide binding affinity with HLA alleles pan-specifically.
DeepTAP
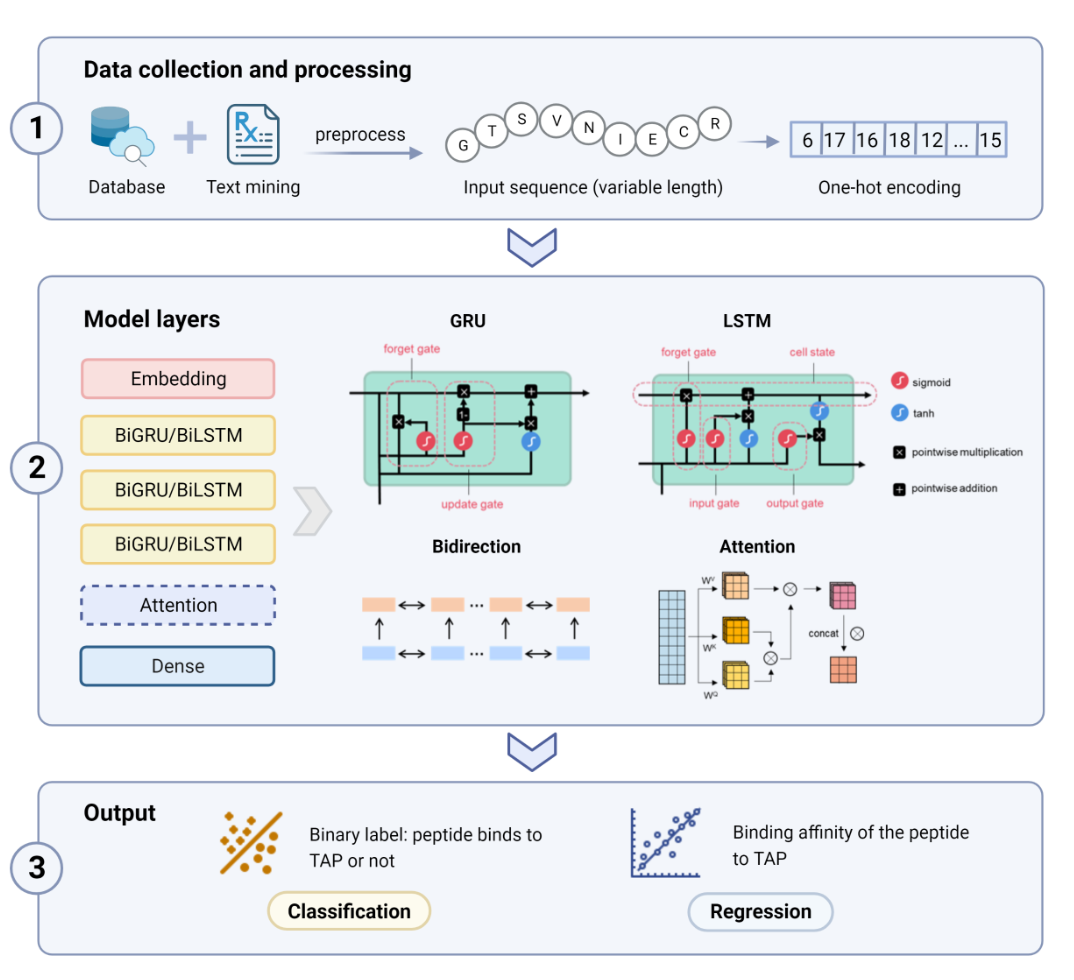
Description:
DeepTAP is a tool based on recurrent neural network(RNN) to predict the binding relationship and binding affinity between antigen associated transporter (TAP) and 6-17mer peptides.
Publication(s):
1. Xue Zhang, Jingcheng Wu, Joseph Baeza, Katie Gu, Yichun Zheng*, Shuqing Chen*, Zhan Zhou*. DeepTAP: An RNN-based method of TAP-binding peptide prediction in the selection of tumor neoantigens. Comput Biol Med. 2023 Jul 8;164:107247.Contact:
Xue Zhang: 22119130@zju.edu.cnURL:
Stand-aloneWeb-server
Spanve
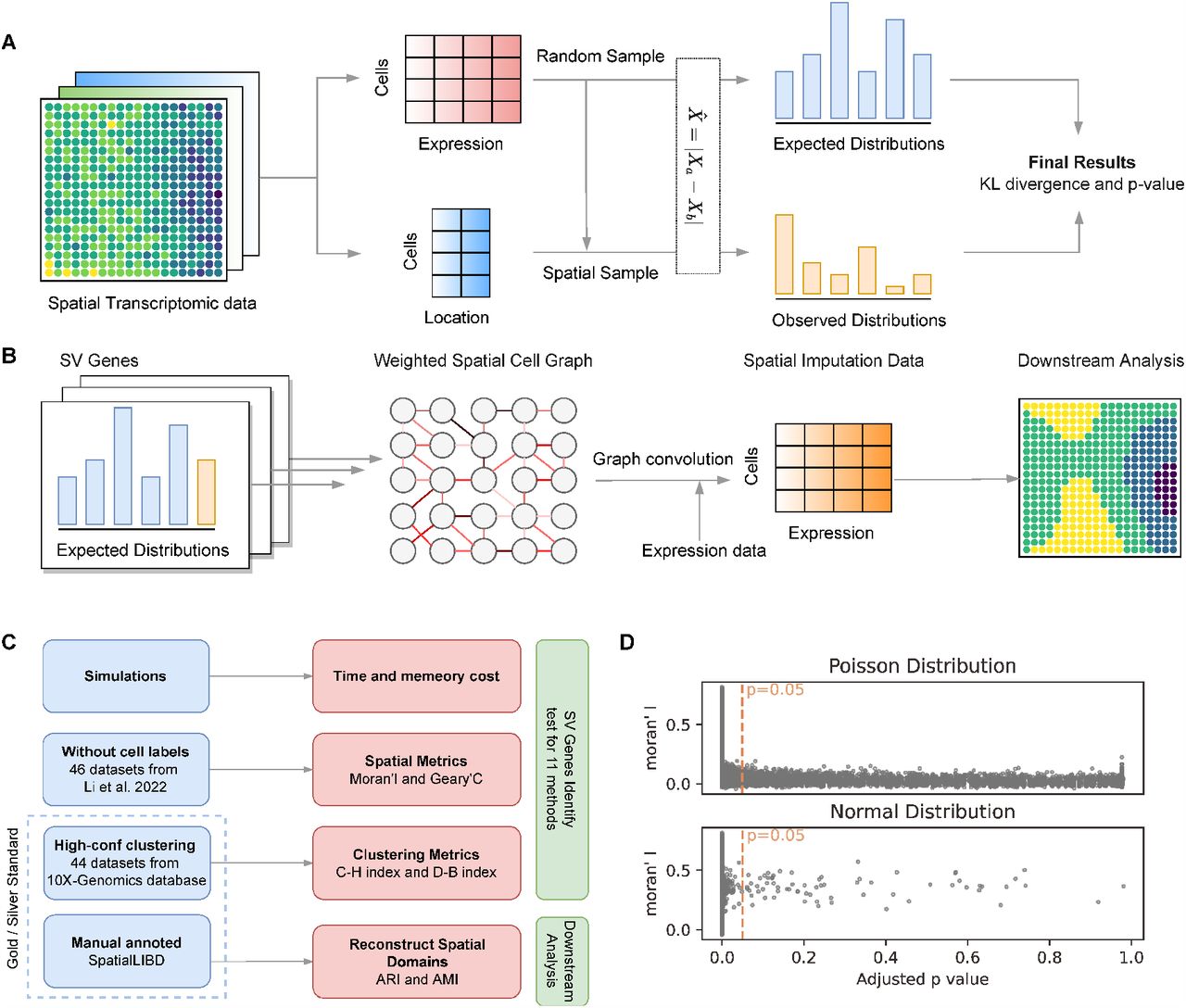
Description:
Spanve, a non-parametric statistical approach based on modeling space dependence as a distance of two distributions for detecting SV genes. The high computing efficiency and accuracy of Spanve is demonstrated through comprehensive benchmarking. Additionally, Spanve can detect clustering-friendly SV genes and spatially variable co-expression, facilitating the identification of spatial tissue domains by an imputation.
MATTE
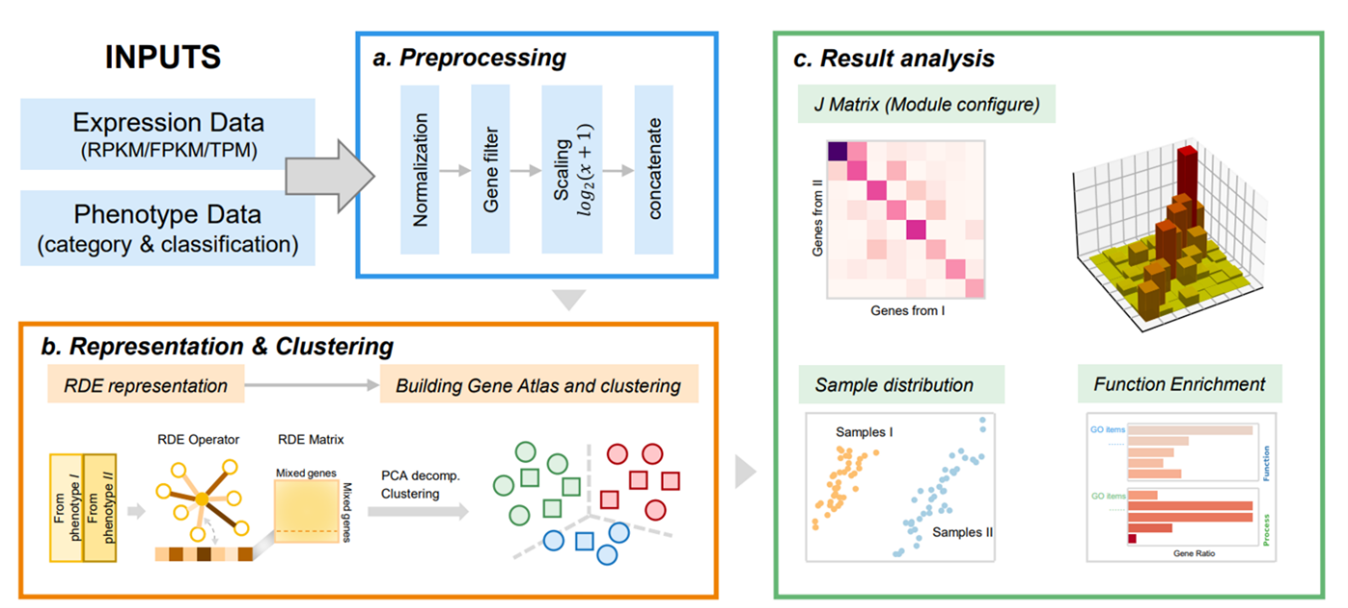
Description:
MATTE (Module Alignment of TranscripTomE) is a python package aiming to analyze transcriptome from samples with different phenotypes in a module view. Differential expression (DE) is commonly used in analyzing transcriptome data. However, genes do not work alone, they collaborate. In recent years, network and module-based differential methods have been developed to obtain more information. New problems appear to make sure module or network structure is preserved in all of the phenotypes. To that end, we proposed MATTE to find the conserved module and diverged module by treating genes from different phenotypes as individual ones. By doing so, meaningful markers and modules can be found to understand the difference between phenotypes better..
TEMPO
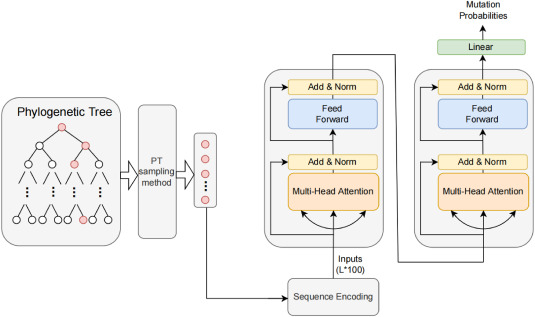
Description:
We design a A transformer-based mutation prediction framework for SARS-CoV-2 evolution called TEMPO. TEMPO is effective for mutation prediction of SARS- CoV-2 evolution and outperforms several state-of-the-art baseline methods. We further perform mutation prediction experiments of other infectious viruses, to explore the feasibility and robustness of TEMPO, and experimental results verify its superiority.
DeepCIP
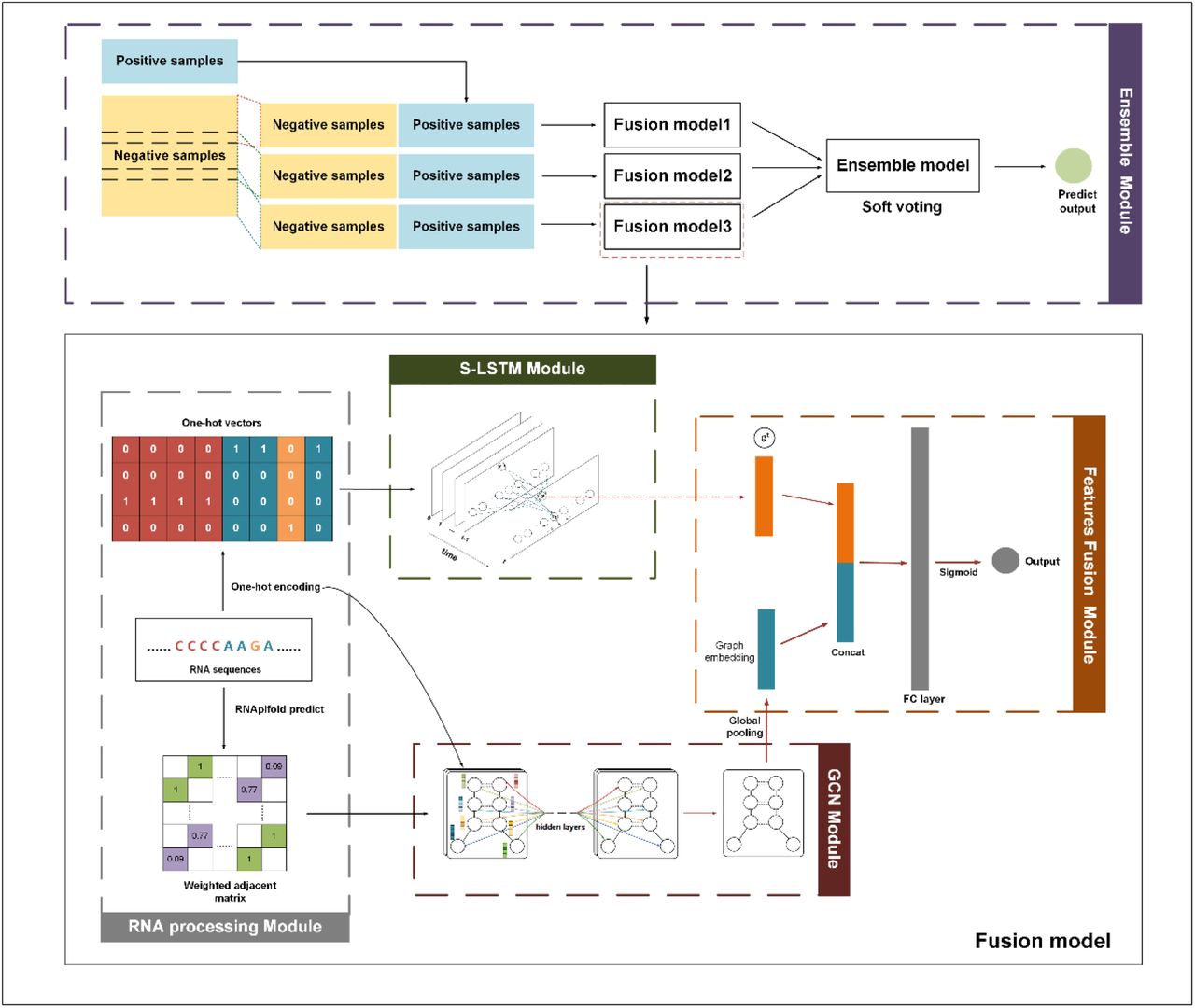
Description:
we proposed DeepCIP, a multimodal deep learning approach for circRNA IRES prediction, by exploiting both sequence and structure information. As far as we know, DeepCIP is the first predictor for circRNA IRESs, which consists of an RNA processing module, an S-LSTM module, a GCN module, a feature fusion module, and an ensemble module. The comparative studies show that DeepCIP outperforms other comparative methods and justify the effectiveness of the sequence model and structure model of DeepCIP for extracting features.
MODIG
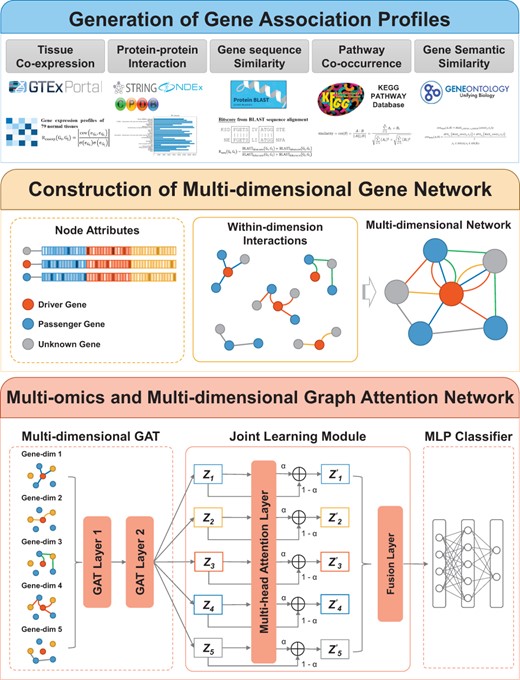
Description:
MODIG is a GAT-based model designed for generating gene repre-sentation from a multi-dimensional gene network for the identifica-tion of cancer driver genes, which mainly performed three steps to integrate multi-omics data and multiple gene associations. Firstly, it generates multiple gene association profiles based on PPI, gene sequence similarity, gene tissue-specific expression profiles, gene pathway membership, and GO annotation information. Secondly, a multi-dimensional gene network is constructed using these gene association profiles as multiple edges and multi-omics features as node attributes. Thirdly, to learn knowledge from the multi-dimensional graph, instead of fusing different edges into a single edge to form a homogeneous graph, M ODIG applies a GAT block for within-dimension interactions to get the dimension-specific gene representations and a joint learning module to adapta-tively learn the importance of different dimensional representa-tions and fuse them by an attention mechanism for downstream cancer driver gene prediction. By doing so, meaningful markers and modules can be found to understand the difference between phenotypes better..
Database
TSNAdb
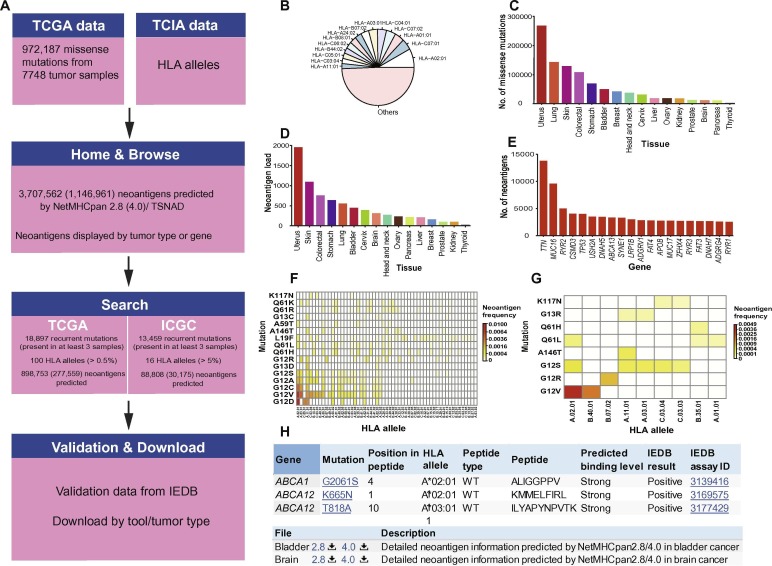
Description:
Tumor-Specific NeoAntigens have attracted more attentions for their importance to cancer diagnosis, prognosis and targeted therapy, as they are crucial tumor biomarkers in identifying tumor cells and are potential targets for cancer immunotherapy. We analysed whole genome/exome sequencing data of 9,155 patients in International Cancer Genome Consortium database (ICGC) and predicted tumor-specific neoantigens including excellular mutations of membrane proteins and neoantigens presented by class I Major Histocompatibility Complex (MHC) molecules. We mapped all the missense mutations to the extracellular regions of membrane proteins and got a dataset contains 88,354 extracellular mutations. We used software NetMHCpan (v2.8) to predict the affinity between Human Leukocyte Antigen (HLA) and collected peptides and obtained a large amount of records with respect to binding and specific binding information.
Publication(s):
1. Wu J, Zhao W, Zhou B, Su Z, Gu X, Zhou Z*, Chen S*. TSNAdb: a database for tumor-specific neoantigens from immunogenomics data analysis. Genomics Proteomics Bioinformatics. 2018, 16(4),276–282.2. Wu J, Chen W, Zhou Y, Chi Y, Hua X, Wu J, Gu X, Chen S, Zhou Z*. TSNAdb v2.0: The Updated Version of Tumor-specific Neoantigen Database. Genomics Proteomics Bioinformatics. 2023, 21.
CandirsDB
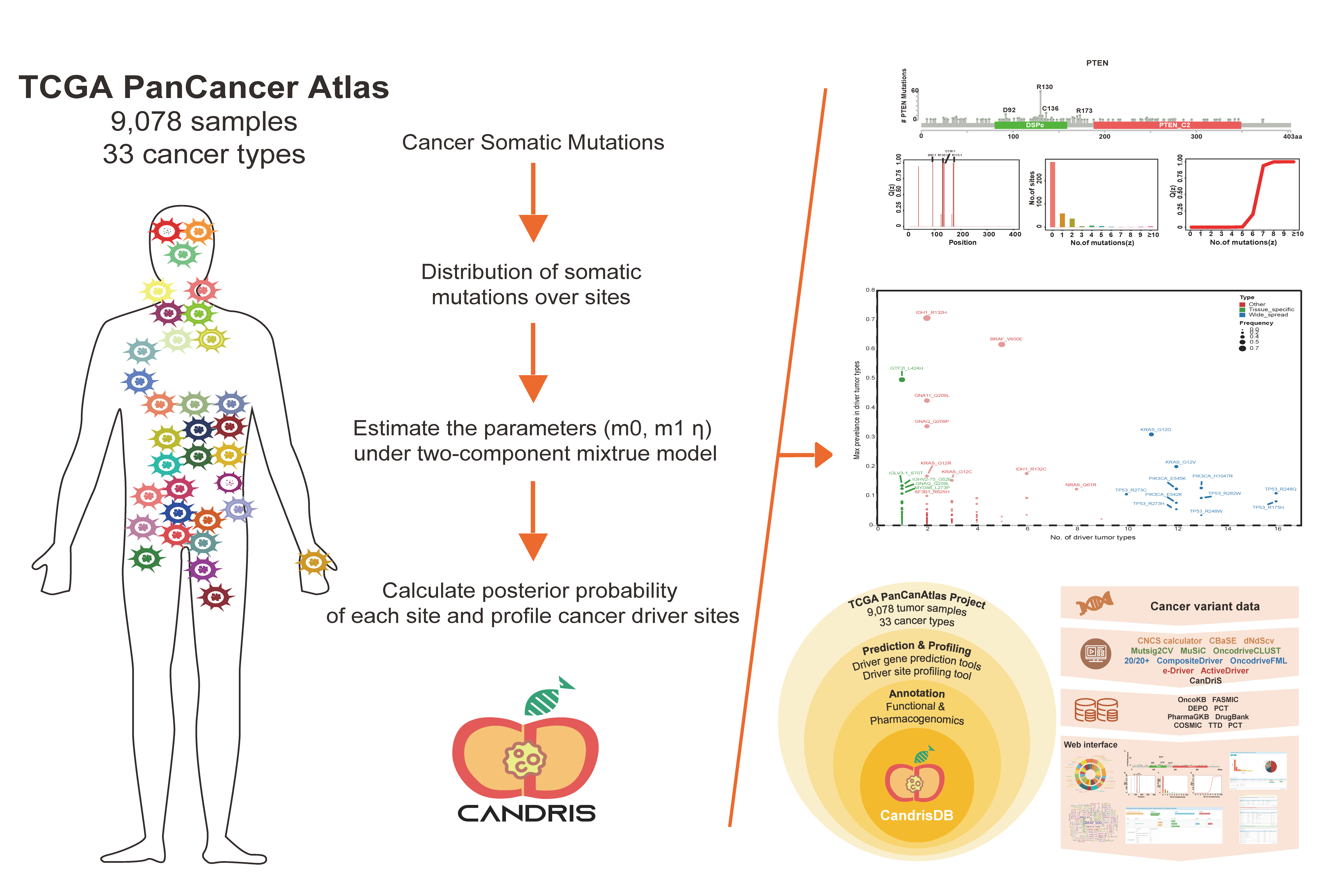
Description:
CandrisDB is a platform to comprehensively proflie the cancer-driving sites at the pan-cancer and tumor-type level for the somatic mutations collected from The Cancer Genome Atlas (TCGA PanCanAtlas project) and International Cancer Genome Consortium (ICGC) (ICGC Release 25) by an in-house method CanDriS. CandrisDB also combined the lists of known driver genes and the predicted results of published bioinformatics algorithms (an in-house method CNCS calculator and other ten algorithms) to compile a list of candidate dirver genes. We also collected data from other public databases on functional and pharmacogenomics annotation for the cancer-driving sites, to provide guidance on clinical medication in the upcoming era of Precision Medicine.
COVIEdb
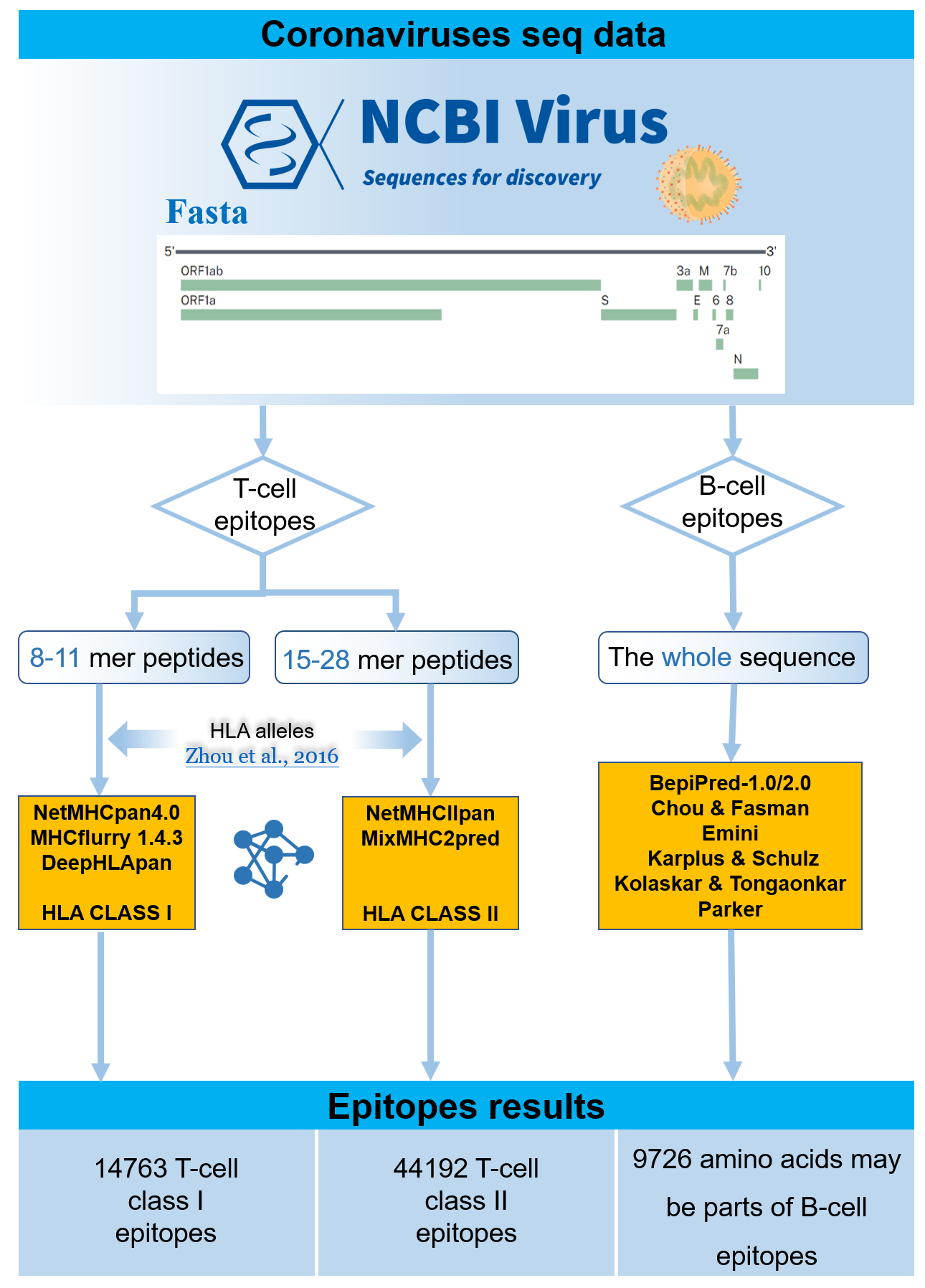
Description:
We predict all the potential B/T cell epitopes for 2019-nCoV, RaTG13-CoV, SARS-CoV and MERS-CoV based on the proteins they express for providing potential targets of vaccines that could effective to different coronaviruses. RaTG13-CoV is added because of its high homology with 2019-nCoV (96% whole genome identity).
Publication(s):
1. Wu J, Chen W, Zhou J, Zhao W, Sun Y, Zhu H, Yao P, Chen S, Jiang J*, Zhou Z*, COVIEdb: A database for potential immune epitopes of coronaviruses, Front Pharmacol, 2020, 11:572249.Contact:
Jingcheng Wu: 11819049@zju.edu.cnURL:
https://pgx.zju.edu.cn/coviedb/CovEpiAb
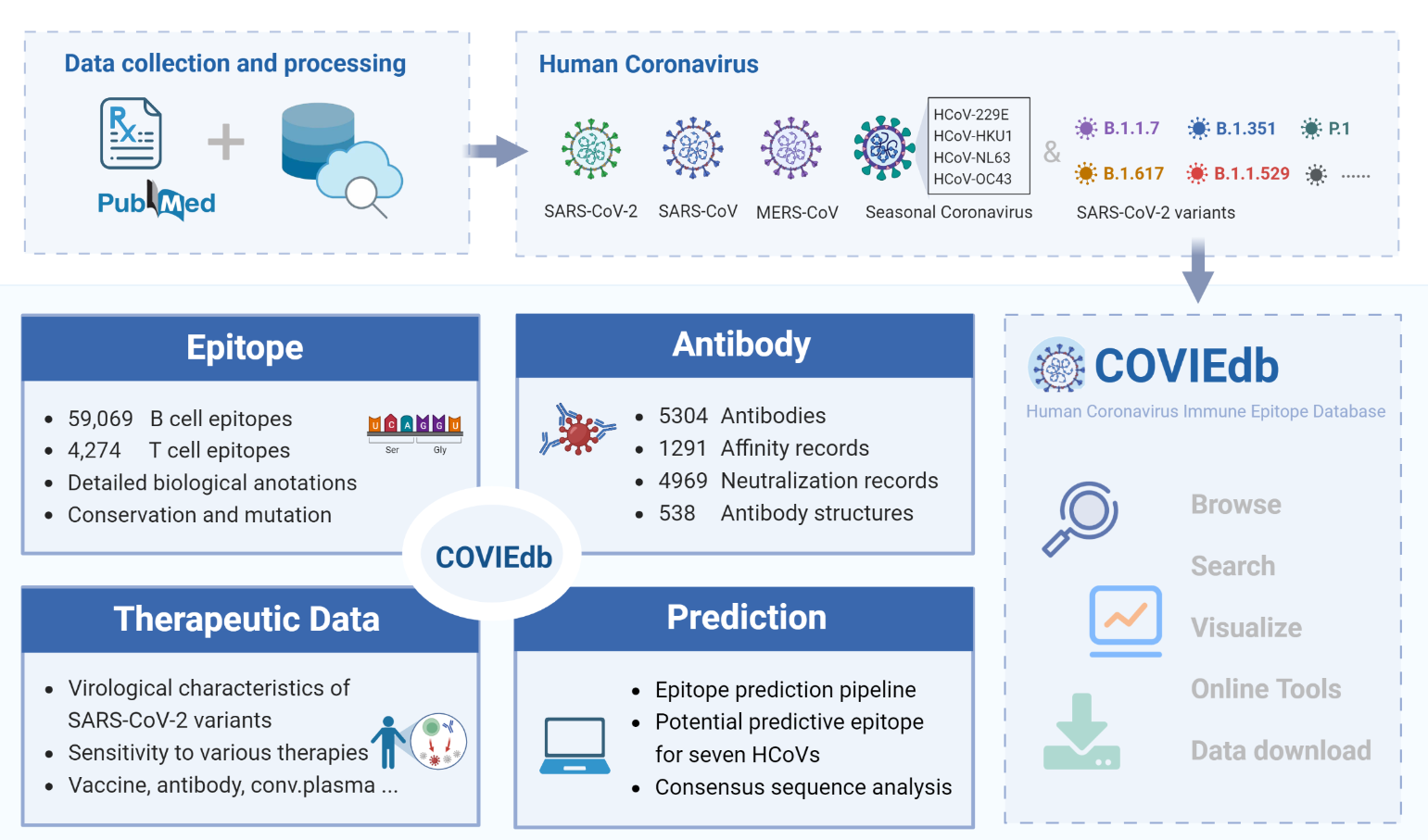
Description:
What sets COVIEdb 2.0 apart from version 1.0 is its systematic collection of diverse immune epitopes and antibodies specific to human coronaviruses (HCoVs). The database includes extensive biological annotations and interaction profiles, providing a comprehensive understanding of these immune responses. Researchers and scientists can rely on this wealth of information to gain insights into the virological characteristics of HCoVs and evaluate the therapeutic efficacy of existing treatments for SARS-CoV-2 variants of concern. To facilitate the identification of high-confidence candidate immune epitopes, COVIEdb 2.0 also offers an integrated tool for immune epitope prediction. This powerful feature, combined with conservation analysis, enhances the efficiency and accuracy of epitope discovery.
Publication(s):
1. UnpublishedOncoTriMD
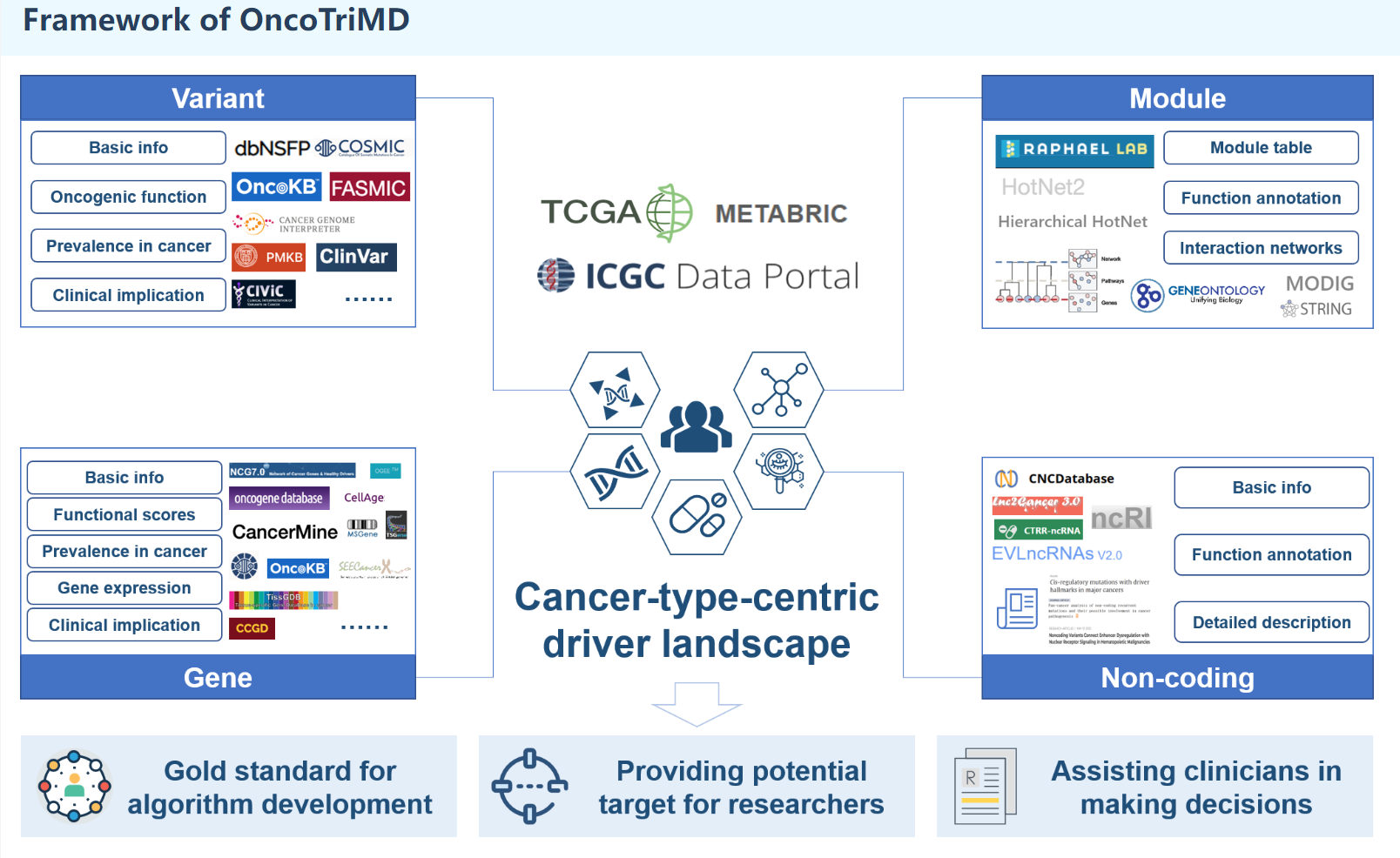
Description:
OncoTriMD is an integrated database that enables streamlined and simultaneous searches for multi-scale cancer drivers. OncoTriMD facilitates the exploration and analysis of various facets of cancer drivers, including driver mutations, driver genes, driver modules, and non-coding drivers. It leverages diverse data sources, including genomic, transcriptomic, and functional impact data, to establish a centralized repository of information on multi-scale oncogenic drivers in various cancer types. Except for non-coding, which is collected manually, all the drivers at the pan-cancer and tumor-type level for the somatic mutations collected from The Cancer Genome Atlas (TCGA PanCanAtlas project), International Cancer Genome Consortium (ICGC Release 28) and METABRIC (Molecular Taxonomy of Breast Cancer International Consortium).
Publication(s):
1. UnpublishedTRAIT
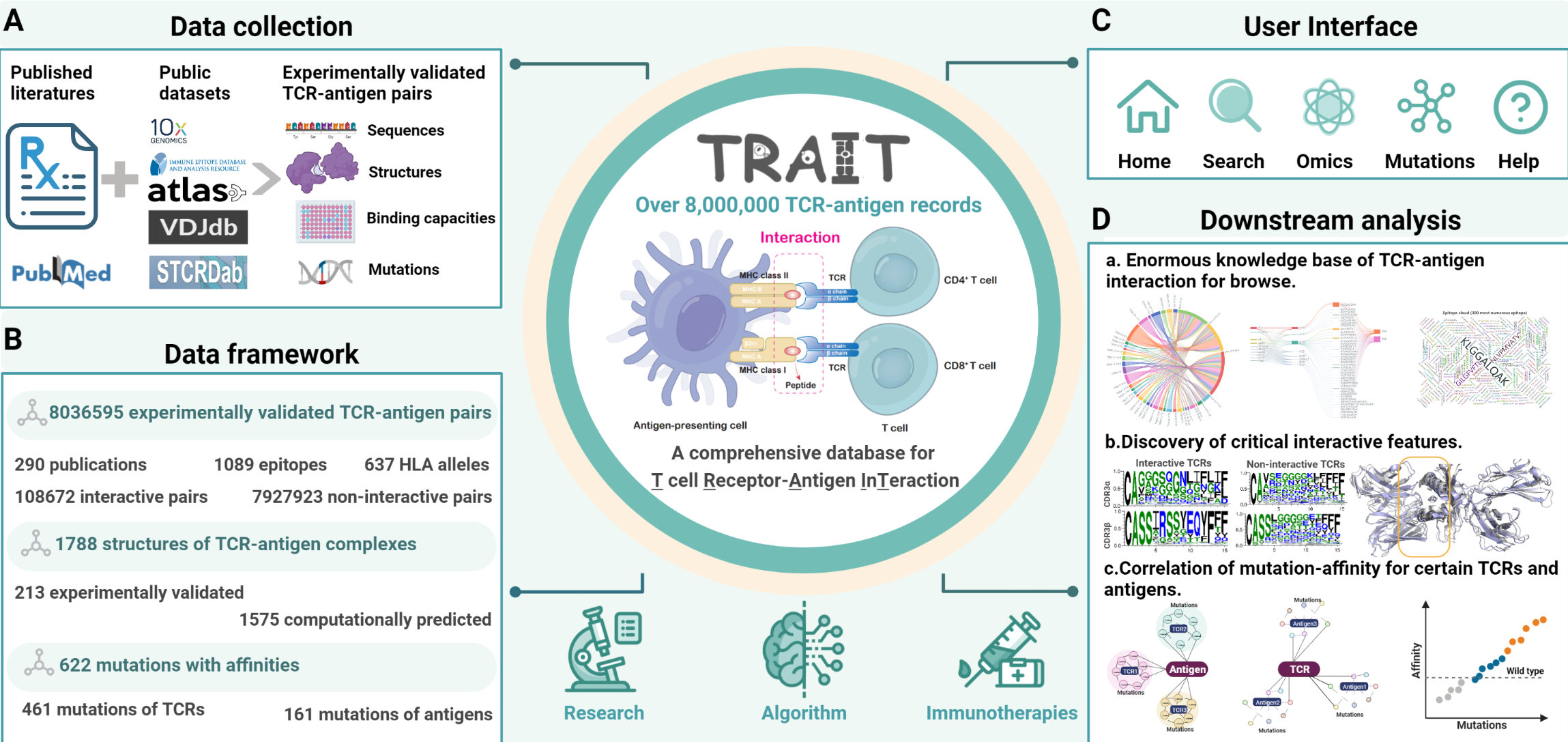
Description:
TRAIT provides landscape of experimentally verified interactive and non-interactive TCR-antigen pairs, together with multi-dimensional perspectives of interaction features integrating sequences, structures and affinities. The unique features of TRAIT include: i) comprehensive and multi-dimensional information of interactive TCR/antigen pairs including sequences, structures and affinities were collected. ii) The most complete structures of TCR/antigen pairs including 213 experimental validated and 1,575 predicted ones as a supplement are focused on to reveal their interface features. iii) Millions of reliable non-interactive TCR/antigen pairs were uniquely collected, allowing users to query features of amino acids that critical for interaction. iv) 622 mutants of TCRs/antigens were displayed on a separated page to dissect traits of their specific recognition and benefit directed evolution of engineered TCRs.
Publication(s):
1. Unpublished(●'◡'●)ノ This website has running: 0 d 0 h 55 min 52 s